Computational Sport
A Formula 1 car is built from about 14,500 individual parts. Each one is designed on a computer, simulated, and then built in a factory. By modeling cars on silicon, before building them in the real-world, F1 teams save money and (ideally) stay below their $135 million cost-cap.
The simulation and modeling tools used by F1 teams are incredible, in part, because they span every scale. CAD (computer-aided design) tools are used to design a wing, for example, whereas CFD (computational fluid dynamics) tools predict the aerodynamic properties of that wing.
Racing teams simulate every screw, every system, and then the entire car as it races around a track at 220+ miles per hour. Simulators can anticipate the specific lap that a driver will need to come in for a pit stop, due to tire wear. Teams even simulate how other teams will fare in a race, based on their prior performances. F1 racing is a computational sport.
Biology, of course, is not F1. Living cells are often described as “wet,” “messy,” or “unpredictable.” Most mathematical models that attempt to predict biology operate at a single level, and don’t extend much beyond. Scientists may model a lone genetic circuit, or perhaps the growth rate of a particular type of cell. But there is no unifying model that integrates everything, from a cell’s genome sequence all the way up to its behavior in a bioreactor, for mammalian cells. (Markus Covert’s group at Stanford University is building multi-scale cell physiology models for bacteria.)
In a prior blog post, we described our software tool, called Kernel, that can help design DNA constructs in silico. It’s analogous to how F1 teams use CAD tools to design a wing or engine. In this blog post, we reveal our suite of simulators that can predict how engineered cells will perform when cultured in a bioreactor. This is more analogous to the CFD tools that F1 teams use to simulate a wing’s performance on-track.
Our long-term goal is to bring all these tools together — Kernel and the simulators — into a holistic platform to model biological systems. And there’s a good reason for that.
Consider what happens when a scientist engineers a cell to make a medicine. They first design a plasmid, a loop of DNA, that contains all the genes needed to make that medicine. They then put that plasmid into the cells, and the cells make the medicine. Some cells do this better than others. The scientist removes the “losers” and selects the “winners,” the cells that make the most medicine. The winners are then placed into a bioreactor, and the scientist tunes and tweaks the bioreactor conditions until hitting upon the “optimal” conditions for that specific cell.
In other words, the conditions for each step in a bioprocess are dependent upon the conditions that came before. The conditions of a bioreactor are based on the characteristics of the “winning” cell. And the “winning” cell is based on the characteristics of the plasmid that went into it. This means that each “optimization” is biased by the step that preceded it.
A Formula 1 team would never design an engine, simulate it, fabricate it, and then build the rest of the car around it. They instead design the wing, the engine, and the suspension all together, holistically, to get the best performance out of the entire car. We should do the same for biology.
CHO Simulator
Chinese Hamster Ovary, or CHO cells, make roughly 70 percent of all F.D.A. approved biologics sold on the market. This includes bestselling antibodies like Humira®, whose sales exceeded $21 billion in 2022, and Keytruda®, a cancer therapy with $20.9 billion sold in the same year.
There is a long history of mathematical models designed to make CHO cells better at making medicines. Indeed, our own simulator stands on the shoulders of giants, adapting insights from prior work developed by researchers in Ireland and the U.K. Scientists at Pfizer and elsewhere have similarly built simulators to predict CHO cell metabolism, and entire companies have spun up to enhance biologics manufacturing using data-driven CHO metabolism models.
Our CHO simulator is built from smaller, interlocking models that, together, predict how a cell’s metabolism will shift during the entire course of a bioreactor experiment. The model can identify the best way to culture cells, including what to feed them (and when), temperature, and agitation rates to boost their medicine-making potential.
We use a dynamic flux-balance analysis model, or dFBA, to predict how metabolites will change over time. A dFBA model essentially “looks” at a cell’s genome, uses algorithms to determine which enzymes the cell expresses, and then anticipates how those enzymes will interconvert molecules in the cell. It considers the concentrations of metabolites outside the cell, and supposes the cell has a particular objective it’s trying to fulfill, such as: “Grow as quickly as possible!” or “Make as much medicine as possible!”
A dFBA model can predict, for example, how quickly a sugar molecule will break down, and how the carbons from that sugar molecule will wend their way through a cell’s metabolic pathways. These models can also predict how the extracellular concentrations of molecules will change over the time of a process, capturing how metabolism shifts in response to a dynamic environment.
.png)
There are a couple limitations to traditional dFBA models, however; they suppose the cell’s objective doesn’t change over time, and their awareness of the conditions in the bioreactor is limited. To address these limitations, we’ve wrapped our dFBA model in a phenotype, or cell-state, model. This “outside” layer is made from a combination of artificial neural networks and ordinary differential equations that consider not only the environment of the bioreactor – such as the concentrations of metabolites within it – but also the internal states of the cells, including which enzymes are likely to be expressed.
By layering the two models, we are able to connect the external variables in a bioreactor to the internal dynamics of a living cell. And the two layers communicate both ways: Outputs calculated by the dFBA are continuously fed back into the phenotype model, and vice versa, in a closed loop of computational calculations.
Our model also has some unique features that make it suitable for holistic optimizations, ranging from genetic design all the way up through a cell’s performance in a bioreactor. The simulator is built upon a “right-sized” metabolic model, for example, that includes all the most important pathways without being too complicated. The model is also “aware” of more details of the bioreactor environment than your typical model. The “outside layer,” or cell-state model, can also represent how big the cells are, whether they’re growing, and what they want to eat. We’re now working to combine the CHO simulator with a model for gene expression, so that we can predict how a specific genetic design — say, a plasmid carrying a DNA sequence, added to a cell — will affect how that cell grows and performs in a bioreactor. More on this in a future post.
We’re only able to build these models because we work with an excellent team of synthetic biologists who have devised methods to measure many variables, in real bioprocesses and for a large variety of cell lines, at our Boston laboratories. Our bioreactors are equipped with dozens of sensors that enable us to monitor cells in real-time, collect data, and transmit it to our biostatistics pipelines.
Now that we’ve got our engine modeled, and we’ve started integrating it with the other components of our system, let’s see how we use this model to actually predict how much medicine a cell will be able to make.
On-Track Performance
The complete CHO model predicts the rate of production and rate of consumption of various metabolites, including amino acids, sugar, and lactate. It also predicts titers for the actual medicine (such as an antibody) and the growth rate of the cell population.
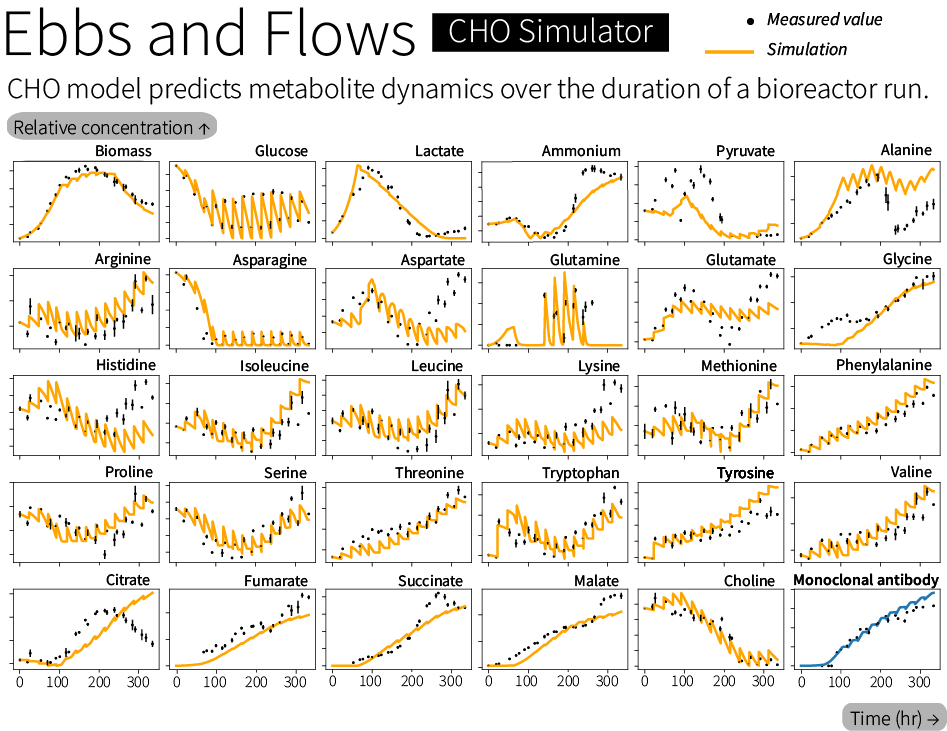
Our predicted results are quite good. We achieved an error of roughly the same as the day-to-day variation in the data across the 31 variables over a 14-day bioreactor experiment. (For those with a statistical bent, that is to say we achieved a mean absolute scaled error of 1.3. A mean absolute scaled error value is expressed relative to the sum of the changes between each measurement and the next.) Between the low error and the wide range of metabolites we predict, this puts our model in good company amongst the best-performing models published in the literature.
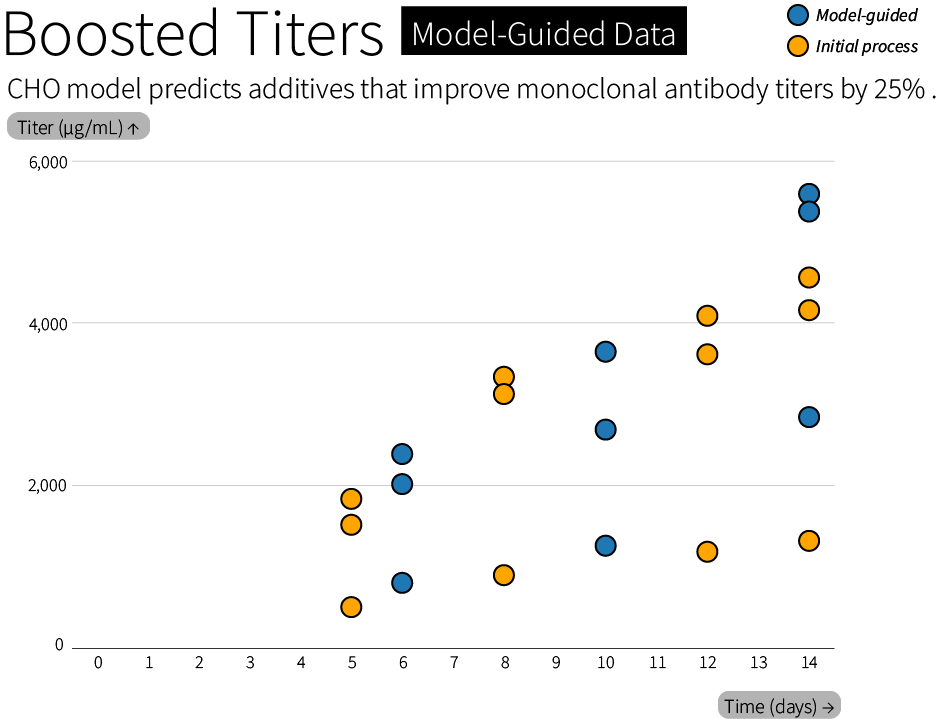
The simulator can reveal metabolic tradeoffs in CHO cells, and we’re already using it to select additives (literally, nutrients added to a bioreactor) that can boost antibody production by 25%. Our first simulations of optimal feeding strategies also identified osmolality, or the concentration of dissolved particles, as a factor that needs to be controlled early on in a bioreactor experiment. The model is already being used to untangle the complex tradeoff between oxidizing and reducing reactions, nitrogen metabolism, and carbon metabolism that limit growth and productivity in a cell culture.
The CHO simulator model will soon be integrated into Kernel, our computer-aided design software for biology, where it will join other tools for plasmid engineering, gene therapy, and more. You’ll be able to use it there, and refine the model’s parameters by inputting your own datasets.
If this work sounds exciting, come join us. Or you can sign up for regular updates below...